
️摩熵化学的科研动态旨在解说当前最热门、最新的期刊内容,我们主要关注有机化学领域,深入探讨物质合成及相关研究的历程与发展。通过每周科研,为您提供有价值的信息和深入的洞察力,帮助您更好地了解有机及其相关领域的发展方向和未来趋势。
️01 通过原子互嵌策略将四个芳香环融合形成四六元环体系
️期刊:Journal of the American Chemical Society
️单位:厦门大学化学化工学院
️作者:Fei-Hu Cui, Le-Han Gao, Kaidong Ruan, Fei Li, Meng Meng, Kexin Ma, Zhengyu Lu, Jiawei Fei, Huayu Tian, Liu Leo Liu, Yu-Mei Lin, Haiping Xia
️原文篇名:Fusion of Four Aromatic Rings via an Atom-Mutual-Embedding Strategy to Form a Tetrahexacyclic System
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c00697
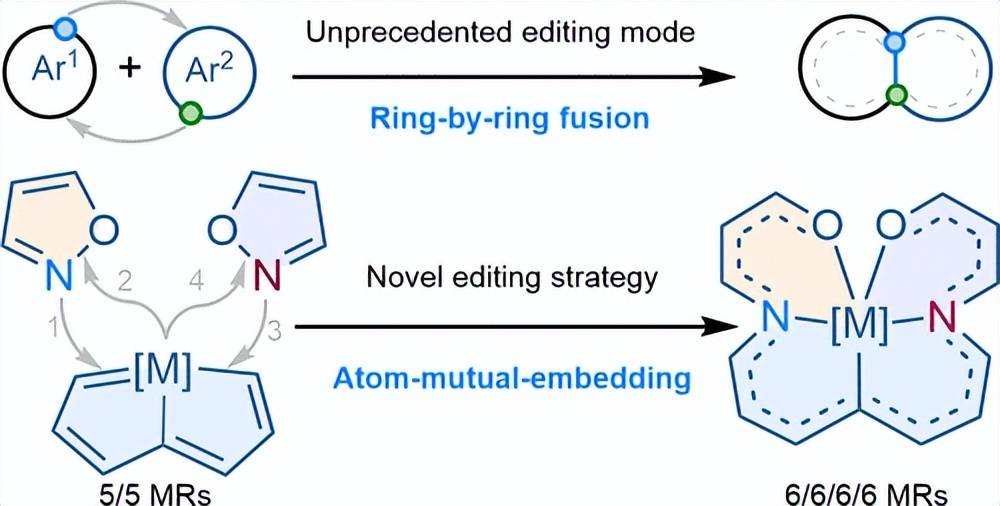
芳香族化合物的骨架操作已经成为合成化学中的有效工具,但由于环和位点选择性的固有复杂性,同时多环操作仍然在很大程度上未被探索。在这里,我们报道了一种前所未有的多环骨架操作,将四个五元芳香环,包括两个有机和两个含金属的芳香体系,融合到一个新的金属桥型6 / 6 / 6 / 6元环骨架中。顺序环融合通过原子互嵌策略完成;该策略包括将两个氮原子逐步插入到单独的金属-碳键中,同时将一个金属原子作为跨越两个异恶唑部分的桥梁。中心金属原子的存在对于确保精确的底物对齐以及增强环和位点特异性至关重要。所得的四六环产物表现出显著的稳定性和优异的近红外( NIR )功能特性,超过了前体化合物。这项工作不仅为设计可进行复杂编辑的通用底物分子奠定了概念基础,而且有助于对分子结构进行合理和有针对性的操作。
️02 操纵芳香性以重定向拓扑化学聚合途径
️期刊:Journal of the American Chemical Society
️单位:劳伦斯伯克利国家实验室
️作者:Qingsong Zhang, Zhipeng Pei, Ah-Young Song, Miao Qi, Rebecca Shu Hui Khoo, Chongqing Yang, Tao Xia, Chen Zhou, Haiyan Mao, Zhiyuan Huang, Shiqi Lai, Yunfei Wang, Liang Z Tan, Jeffrey A Reimer, Jian Zhang, Michelle L Coote, Yi Liu
️原文篇名:Manipulating Aromaticity to Redirect Topochemical Polymerization Pathways
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c03077
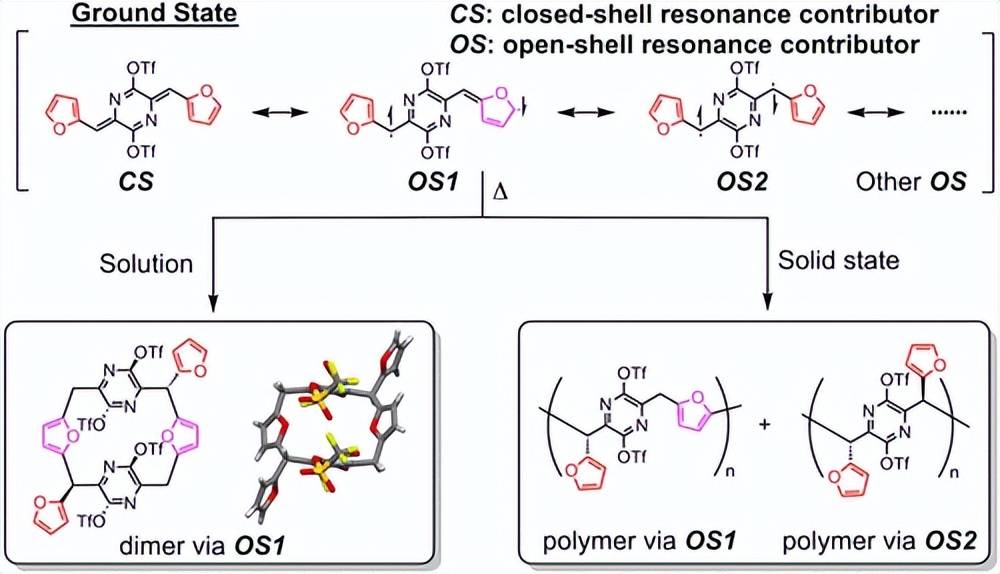
拓扑化学聚合( TCP )是通过固态转化产生区域和立构有规聚合物的重要途径。在此,我们提出了一种通过调节对位氮杂喹二甲烷( AQM )环体系中末端基团芳香性来控制拓扑化学聚合路径的创新策略。用较少的芳香呋喃单元取代苯基基团,在热激活时扩展了共轭核的显著自旋密度离域,在呋喃位置诱导了显著的双自由基特征,并在溶液和固体状态下实现了非传统的反应活性。在甲苯中热处理产生了独特的呋喃-次甲基C - C偶联形成的环番二聚体,X射线晶体学证实了这一点,而固相反应则产生了柱间的呋喃-次甲基偶联和柱内次甲基-次甲基偶联形成的聚合物。通过理论模拟和同位素标记实验验证了这些转变背后的自旋中心导向机制。这项研究突出了功能性亲芳香体系中芳香性调节的能力,这使得合成具有主链结构的聚合物成为可能。
️03 使用 Isayama–Mukaiyama 过氧化的氢脱烷基化 C(sp3)–C(sp2) 键碎裂
️期刊:Journal of the American Chemical Society
️单位:加州大学洛杉矶分校化学与生物化学系
️作者:Jeremy H Dworkin, Zhuoxi M Chen, Kathleen C Cheasty, Aris V Rubio, Ohyun Kwon
️原文篇名:Hydrodealkenylative C(sp3)–C(sp2) Bond Fragmentation Using Isayama–Mukaiyama Peroxidation
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c00540
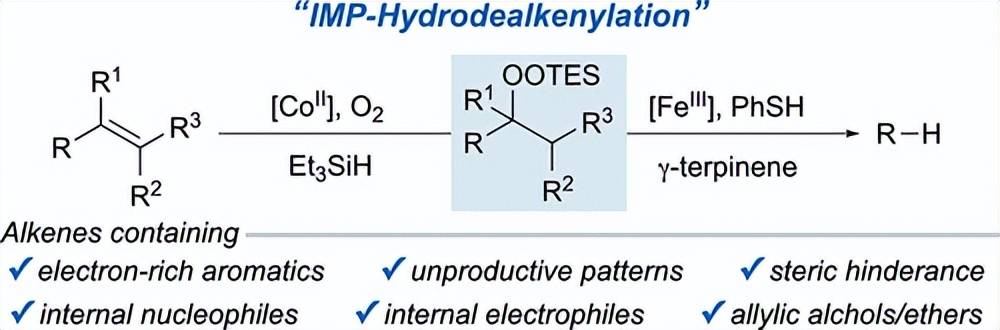
自由基捕获策略的进步扩大了通过脱烯基化合成从烯烃获得的产物范围。然而,这些方法仍然是有限的,因为它们依赖于臭氧分解从烯烃生成关键的过氧化物中间体。臭氧氧化法有几个局限性。与含富电子芳香烃的烯烃不相容。在脱烯基化合成的背景下,它也不适用于某些烯烃取代模式。此外,它还与空间位阻较大的烯烃、内部亲核试剂和亲电试剂以及烯丙醇进行竞争。在本论文中,我们利用Isayama-Mukaiyama 过氧化 (IMP),解决了臭氧化反应在拯救以前难以接近的烯烃底物方面的局限性,拓宽了脱烯基官能团化反应的适用性。特别地,我们将IMP应用于加氢脱烯反应中,并描述了一种新的自由基加氢条件- -在回流的甲醇中使用催化[FeIII]、催化苯硫酚和γ -萜品烯——来解决与IMP生成的烷基硅基过氧化物有关的β断裂问题。
️04 铜催化C - B ( Sp3 )键通过Cu - B ( Sp3 )络合物的中介形成
️期刊:Journal of the American Chemical Society
️单位:香港中文大学化学系
️作者:Zhanqiang Ye, Chun Yin Kwok, Sze Lam Lam, Linlin Wu, Hairong Lyu
️原文篇名:Copper-Catalyzed C–B(sp3) Bond Formation through the Intermediacy of Cu–B(sp3) Complex
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c00974
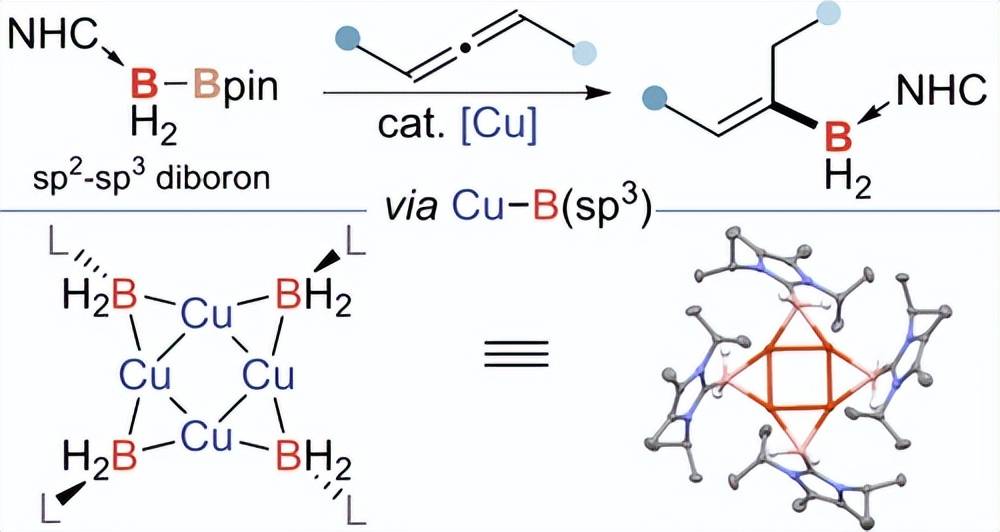
绝大多数过渡金属( TM )催化的硼化转化都依赖于TM-B(sp2)配合物。相比之下,由于缺乏合适的硼(sp3) 试剂,TM-B (sp3)物种的化学及其在催化硼化反应中的潜力仍然令人惊讶。在此,我们利用我们最近开发的sp2-sp3双硼试剂成功实现了铜催化的联烯氢硼化反应构建C-B(sp3)键。包括Cu-B(sp3)配合物的分离和结构表征在内的全面机理研究证实了催化循环中Cu-B(sp3)中间体的存在。
️05 镍催化的不对称合成双嗜性仲膦氧化物
️期刊:Journal of the American Chemical Society
️单位:中国科学技术大学化学系
️作者:Wei-Han Wang, Si-Yu Zhang, Yu-Xiang Zhang, Yin-Qi Wang, Qing-Wei Zhang
️原文篇名:Nickel-Catalyzed Asymmetric Synthesis of Ambiphilic Secondary Phosphine Oxides
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c03897
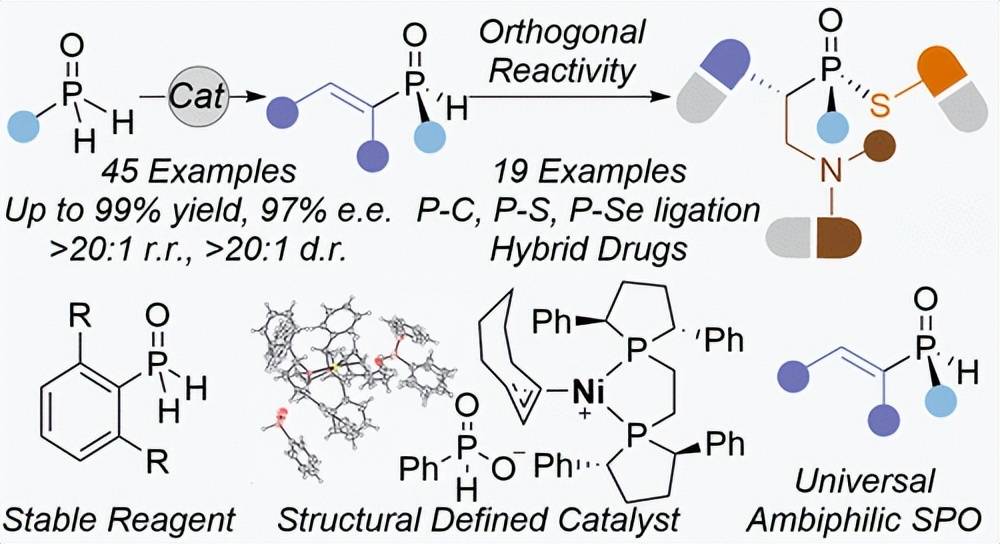
具有强亲核官能团和亲电官能团的双嗜性化合物的合成带来了重大挑战,因为它们容易发生自反应,形成低聚物或聚合物。我们已经成功地利用其有争议的稳定性和反应性,从定制的伯式氧化膦中实现了双亲性 P-立体烯基仲膦氧化物的镍催化不对称合成。该方法对多种未活化的炔烃表现出显著的耐受性,包括来自天然产物和药用相关分子的炔烃,从而为具有高对映选择性和区域选择性的 P-立体膦提供了通用合成子。该模性产物与亲核试剂和亲电试剂均表现出有趣的正交反应性,并且可以很容易地转化为各种医学相关的 P-立体化合物。
️06 通过杂芳基自由基中间体协同光酶催化烯烃抗马尔科夫尼科夫氢芳基化反应
️期刊:Journal of the American Chemical Society
️单位:普林斯顿大学化学系
️作者:Prasun Mukherjee, Zayed Alassad, Todd K Hyster
️原文篇名:Synergistic Photoenzymatic Anti-Markovnikov Hydroarylation of Olefins via Heteroaryl Radical Intermediates
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c01066
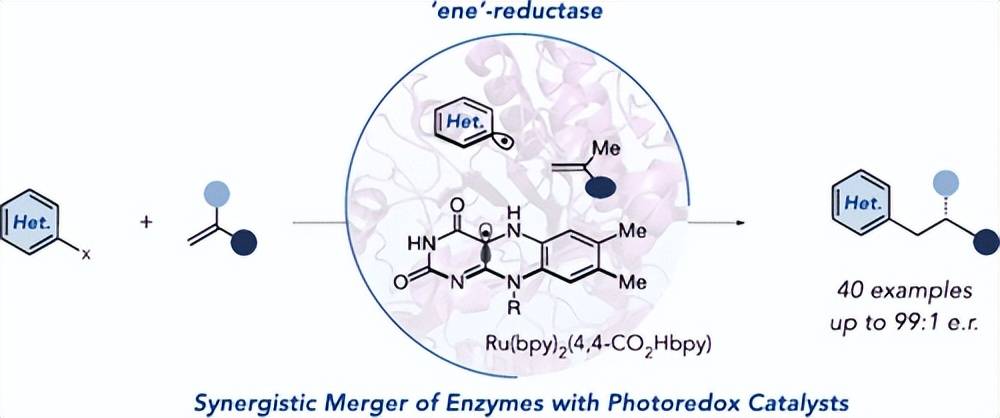
杂芳基烷基化反应是合成生物活性分子不可或缺的反应。使用杂芳基卤化物进行烯烃的抗马尔科夫尼科夫氢芳基化反应,使产物成为单一的区域异构体;然而,催化变体在控制这些反应的立体化学结果方面是无效的。在这里,我们报道了一种使用黄素依赖型' ene ' - reductases和钌光氧化还原催化剂协同光酶催化烯烃的氢芳基化反应。鉴定了酶的同系物,以大于80 %的产率获得了两种产物的对映体,最高可达99:1 er。该方法对苯乙烯基和未活化烯烃均有效,突出了该方法的通用性。产量最高的系统涉及对酶的亲和力增加的羧化光催化剂。该工作拓展了酶可用于立体选择性分子间偶联反应的自由基中间体类型。
️07 对称钴催化氟原子构建四元立体中心
️期刊:Journal of the American Chemical Society
️单位:浙江大学化学系
️作者:Jingyi Wang, Jin Yang, Jian He, Zhan Lu
️原文篇名:Asymmetric Cobalt Catalysis for the Construction of Quaternary Stereogenic Centers with Fluorine Atoms
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c03944
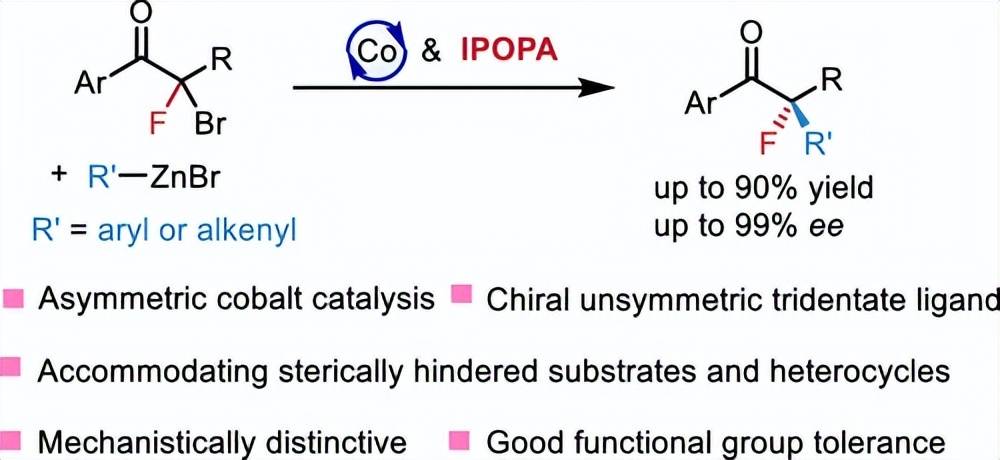
生成氟化季碳手性中心的稳健合成方法学的进步在有机和药物化学领域备受追捧。本研究通过钴催化的α -溴代- α -氟代酮与芳基/烯基锌试剂的不对称Negishi偶联反应成功地提供了此类化合物。手性不对称N,N,N -三齿( CUT )配体的调整在提高钴催化的反应活性和选择性、防止加氢脱溴副产物的生成、空间位阻较大的底物和杂环等方面起到了关键作用。控制和动力学实验表明,转金属化是限速步骤,这一机制特征使得新开发的不对称钴催化交叉偶联不同于以往的方法学。
️08 不对称离子对光氧化还原催化C ( sp3 ) - H芳基化羰基化合物的去消旋化
️期刊:Journal of the American Chemical Society
️单位:中国科学院大学杭州高等研究院
️作者:Chenxi Wen, Zhengke Huang, Sheng-Ye Zhang, Zhimin Li, Bolong Chai, Zheng Huang, Qi-Kai Kang
️原文篇名:Deracemization of C(sp3)–H Arylated Carbonyl Compounds via Asymmetric Ion-Pairing Photoredox Catalysis
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.5c02235
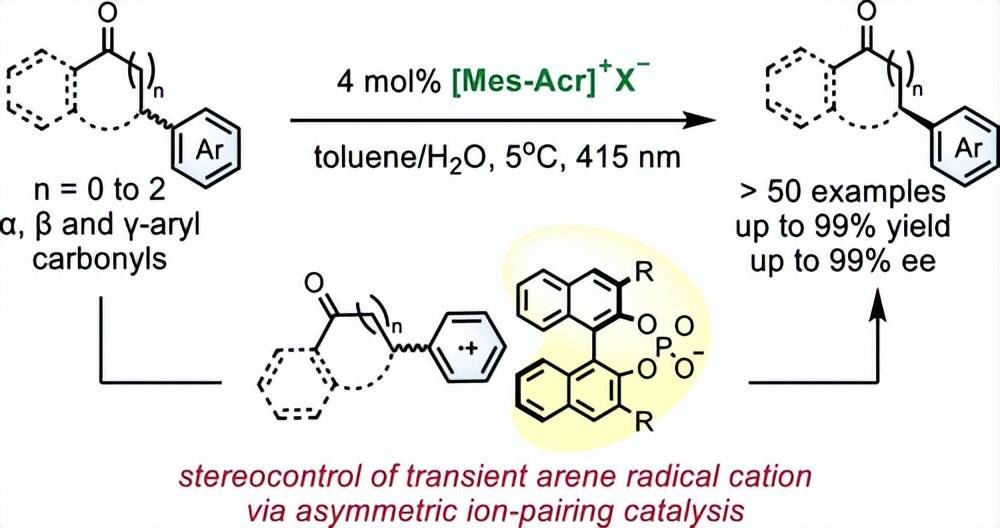
C(sp3)- H芳基化羰基化合物的去消旋化反应面临底物范围的限制。通过芳基的光活化和通过不对称离子对催化对生成的芳烃自由基阳离子的立体控制,我们能够实现在可烯醇化和不可烯醇化的立体中心上芳基化的羰基化合物的去消旋化。各种 α-芳基、β γ-芳基酮和酯,包括天然产物和药物衍生物,可以有效地转化为它们的对映体,具有高对映选择性。通过实验和计算研究相结合的机理研究表明,该反应涉及富电子芳基的单电子氧化,然后通过手性磷酸阴离子对生成的自由基阳离子中间体进行动力学拆分。去质子化被认为是立体决速步骤,而立体选择性的背电子传递和 3 Mes-Acr1+*的三重态猝灭也可能影响光稳态下的对映选择性。
️09 氧化C - H键断裂的末端氢化钒及其在O2还原中的应用
️期刊:Journal of the American Chemical Society
️单位:复旦大学化学系
️作者:Lei Zheng, Yun-Shu Cui, Dong-Ping Chen, Geng-Mu Li, Feng Liu, Dan-Dan Zhai, Zhang-Jie Shi
️原文篇名:Terminal Vanadium Hydride through Oxidative C–H Cleavage and Its Application in Reduction of O2
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.4c14758
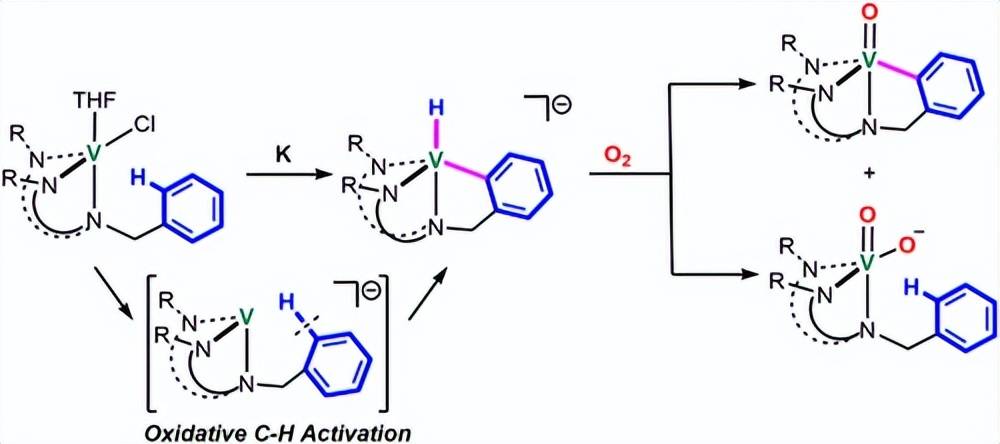
本文报道了一种新型的钒氢化物配合物{ [ DippN2NCH2C6H4 ] VH } 2 { K } 2 ( 2 )的合成和表征,该配合物通过分子内Caryl - H氧化加成反应原位生成低价钒中间体得到。这种钒氢化物表现出强还原性能,能够通过4电子还原过程活化O2。同时,它形成了具有再生Csp2 - H键的二氧钒配合物5和氧钒配合物6。基于氘标记实验和DFT计算,提出了5和6形成的可能机理。
️10 硼试剂在确定吡啶(二卡宾)钴催化氟代芳烃C - H硼化反应位点选择性中的作用
️期刊:Journal of the American Chemical Society
️单位:普林斯顿大学化学系
️作者:Haozheng Li, Hanna H Cramer, Jose B Roque, Carlota Odena, Alex M Shimozono, Paul J Chirik
️原文篇名:The Role of Boron Reagents in Determining the Site-Selectivity of Pyridine(dicarbene) Cobalt-Catalyzed C–H Borylation of Fluorinated Arenes
️原文网址:https://pubs.acs.org/doi/abs/10.1021/jacs.4c15596
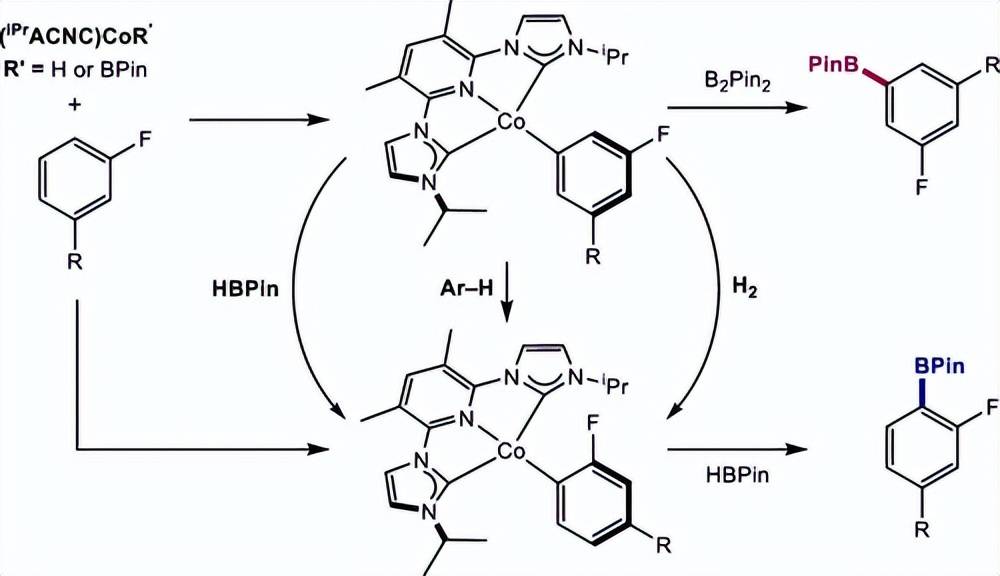
研究了吡啶(双卡宾)钴催化剂促进的氟代芳烃与B2Pin2和HBPin的C(sp2)-H硼化反应中间位和邻位对氟选择性的起源。原位生成的钴(Ⅰ) -硼络合物与3种代表性的氟代芳烃发生间位选择性 C(sp2)-H 氧化加成反应,主要形成相应的钴(Ⅰ) -芳基络合物的间位异构体。尝试观察或分离四配位的钴( I ) -硼基配合物,得到钴氢化物二聚体 [(iPrACNC)CoH]2、硼氢化物 (iPrACNC)CoH2BPin 或氢二硼酯 (iPrACNC)CoH(BPin)2,具体取决于 B2Pin2 的量和 HBPin 存在。制备亚磷酸酯衍生物 (iPrACNC)CoH(P(OiPr)3) 和 (iPrACNC)CoBPin(P(OiPr)3) ,并对其进行了晶体学表征。
在间二氟苯的催化硼化反应中,尽管间氟硼化反应是催化的主要产物,但邻氟钴( I ) -芳基和硼氢化物络合物被确定为静息状态。氘的动力学同位素效应支持不可逆的但不是周转限制的 C(sp2)–H氧化加成。分离得到的钴(Ⅰ) -芳基中间体与B2Pin2的化学计量硼化反应表明,间位钴(Ⅰ) -芳基比邻位异构物具有更高的反应活性,并解释了观察到的钴(Ⅰ) -芳基静息态。所有的钴(Ⅰ) -芳基化合物与HBPin的反应都较快。
当邻位钴(Ⅰ) -芳基化合物以高位点选择性得到芳基硼酸酯产物时,间位钴-芳基化合物得到芳基硼酸酯异构体和自由芳烃的混合物。与DBPin的氘标记实验证实HBPin介导可逆的 C(sp2)–H氧化加成。因此,整体的位点选择性来自两个增强效应:( i )动力学上的亚选择性氧化加成和( ii )间位钴-芳基异构体与B2Pin2的更快反应。由于B2Pin2转化为HBPin, C(sp2)–H还原消除竞争间位钴-芳基异构体的硼化,导致邻位选择性硼化增加。
声明:以上内容仅代表作者观点,如有不科学之处,请在下方留言指正。